Development of an Open-Source Integrated Quantum Chemistry Calculation and Deep Learning Analysis Platform for Predicting and Understanding Polymer miscibility
Development of an Open-Source Integrated Quantum Chemistry Calculation and Deep Learning Analysis Platform for Predicting and Understanding Polymer miscibility
July 13, 2023
A research team from the “ISM-MCC Frontier Materials Design Laboratory”*1, a joint research division of the Mitsubishi Chemical Group*2 (MCG) and the Institute of Statistical Mathematics (ISM), has developed a new method to accurately predict the miscibility of polymer-solvent systems. A paper summarizing the results of this research was published in the journal “Macromolecules ” (American Chemical Society) on July 10, U.S. time. ISM and MCG have developed an integrated quantum chemistry calculation and machine learning platform to accurately and quickly predict interaction parameters (χ [chi] parameters) that represent the miscibility of polymers and solvents. To obtain a highly accurate predictive model from limited experimental data with systematic biases that derive from experimental systems, the research team used large amounts of data from quantum chemistry calculations generated by MCG’s high-performance computer. By training the model with these tasks simultaneously using a technique called multi-task learning, we have succeeded in building a predictive model that is applicable to a wider range of polymers and solvents than conventional machine learning models. Furthermore, the developed model was confirmed to be capable of calculating χ parameters about 40 times faster than conventional methods based on quantum chemistry calculations. The dissolution of polymers in solvents is an essential process used in plastic recycling, as well as in many areas of materials development including polymer synthesis, purification, painting, and coating. As such, the results of this research are expected to contribute toward solving a variety of issues in science and industry. In addition, we have made part of the developed source code and data available to the public free of charge in order to contribute to the promotion of open innovation and open science in the field of materials informatics. *1: https://www.m-chemical.co.jp/news/2019/__icsFiles/afieldfile/2020/02/18/190610-2.pdf |
Background of this Research
The dissolution of polymers in solvents is one of the essential processes used in materials development, including plastic recycling, as well as polymer synthesis, purification, painting, and coating. For example, solvents are added to plastic waste consisting of a mixture of different types of plastics to selectively separate only certain materials. Solvents are also used as “miscibilizers” to create high-performance polymer blends. As such, predicting the miscibility of polymers and solvents is an important challenge for both science and industry.
It is known to be difficult to accurately predict the phase behavior of various polymer-solvent systems using current computational chemical techniques. According to the Flory-Huggins theory*3, which describes the thermodynamic properties of polymer solutions, in instances where temperature, volume fraction, and molecular chain length are given, the free energy of mixing*4 of a polymer solution is determined by a quantity called the χ parameter, which represents the interaction between the polymer and solvent. The most widely used methods for predicting the χ parameter are empirical methods of prediction based on the distance between the solubility parameters of polymers and of solvents. For example, the Hansen solubility parameter (HSP) represents a molecule as a three-dimensional vector consisting of a dispersion term, polarity term, and hydrogen bond term. Polymer-solvent miscibility is estimated based on the distance between HSP vectors. While solubility parameters for many molecules have been measured experimentally, empirical models such as the atomic group contribution method*5 are applied to estimate solubility parameters of molecules whose solubility parameters are unknown. That being said, the prediction accuracy of empirical models such as this is known to be very low outside of specific molecular species. The COSMO-RS method*6 based on quantum chemistry calculation can also be used to estimate χ parameters, but quantum chemistry calculation is time-consuming, making it difficult to apply to, for example, large-scale screening of candidate solvent molecules. Moreover, the prediction accuracy of this method is not high. Furthermore, the training dataset is quantitatively insufficient for building machine learning-based predictive models, and it is known to be significantly biased by the nature of the experimental systems. To resolve these issues, the ISM-MCC Frontier Materials Design Laboratory aimed to build an accurate predictive model applicable to a wide range of polymer-solvent combinations by integrally analyzing large amounts of quantum chemistry calculation data and limited experimental data using a method known as multi-task learning.
Research Details and Results
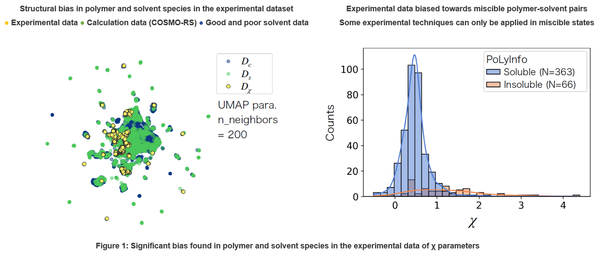
To train the model, experimental values for the χ parameters of 1,190 polymer-solvent pairs, consisting of 46 different polymers and 140 different solvent molecules, were used. The dataset also included χ parameter measurements for different temperatures and compositions. The molecular species of polymers and solvents in the dataset are distributed over a very limited region of the overall chemical space (Figure 1, left). Also, because it is difficult in some experimental systems to measure the χ parameter of polymer-solvent systems in an immiscible state, a large bias exists in the distribution of data (Figure 1, right). As such, models trained using only this dataset will generally have a limited range of applicability for predictions, and will not be able to predict χ parameters for immiscible states.
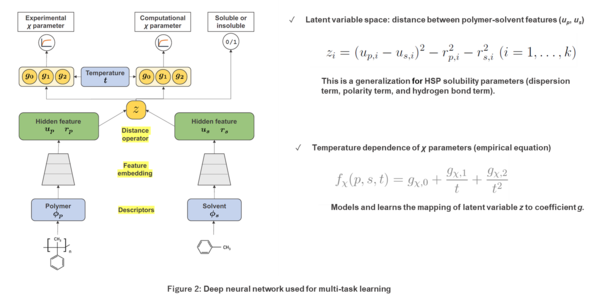
To solve this problem, we used the COSMO-RS method, which employs quantum chemistry calculations, to generate a dataset of the χ parameters of 9,129 polymer-solvent pairs.We also created a dataset with binary labels assigned to 29,777 different polymer-solvent combinations indicating whether the solvent was experimentally found to be a good or poor solvent. Using these three datasets, we trained a deep neural network that predicts experimental χ parameter values, values obtained using the COSMO-RS method, and binary labels indicating the solubility of the polymer and solvent from the chemical structures of the polymer and solvent (Figure 2). This method is called multi-task learning. In multi-task learning, different tasks with a common underlying mechanism are learned simultaneously in a unified model. Experimental data for χ parameters of the primary task are quantitatively limited and contain biases that derive from experimental systems. To address this issue, we defined the two auxiliary tasks and used the data encompassing a wide range of molecular species for training, and by doing so were able to expand the range of applicability of the predictive model. We have also confirmed that the model is capable of calculating χ parameters about 40 times faster than conventional methods based on quantum chemistry calculations.
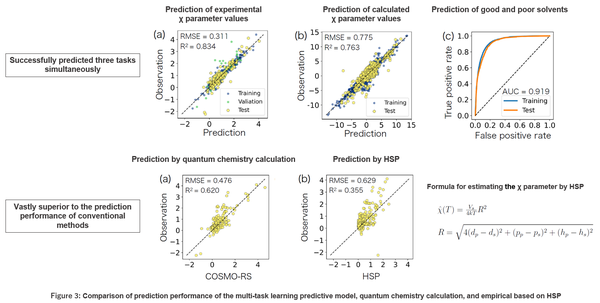
This model was experimentally verified to have very high predictive performance in all three tasks. It also exhibited far greater predictive power than quantum chemistry calculations using the COSMO-RS method or empirical methods based on HSP (Figure 3). The architecture of this model is designed to extend the HSP concept. HSP posits that the potential solubility of a molecule is determined by its dispersive power, polarity, and hydrogen bond strength. Meanwhile, machine learning algorithms have suggested that there are 34 different factors involved in molecular solubility. Some of these factors were found to correspond to the three HSP factors. This suggests that there are unknown factors in the mechanisms for determining polymer-solvent miscibility that have been disregarded in HSP.
Actions going forward
This research has resulted in the completion of an integrated quantum chemistry calculation and deep learning analysis platform for building models to predict the miscibility of polymer-solvent systems. We plan to continue improving the predictive performance of the model as we continue to produce data.
Going forward, predicting and understanding of the miscibility of polymers and solvents will become increasingly important in materials development. In recent years in particular, expectations for technological innovations for recycling waste plastic resources are growing rapidly as we move toward realizing decarbonized societies. The development of miscibilizers for various types of polymers will be essential in improving the recycling rates of waste plastics. ISM-MCC Frontier Materials Design Laboratory will be deploying this model for practical applications in miscibilizer development. In order to further improve and extend machine learning techniques and to promote open innovation and open science in the field of materials informatics, we have made part of the developed source code and data available to the public.
Paper published
Title of paper: Multitask machine learning to predict polymer-solvent miscibility using Flory-Huggins interaction parameters
Authors: Yuta Aoki, Stephen Wu, Teruki Tsurimoto, Yoshihiro Hayashi, Shunya Minami, Okubo Tadamichi, Kazuya Shiratori, Ryo Yoshida
Journal: Macromolecules
DOI: 10.1021/acs.macromol.2c02600
Publication date and time: 9:00 (20:00 U.S. time), July 11, 2023
Terminology
*3: Statistical thermodynamics theory based on a lattice model independently proposed by Flory and Huggins in 1942. To this day, this is often cited in discussions of the thermodynamic properties of polymer solutions and polymer mixtures.
*4: Change in free energy by the mixing of two components.
*5: A method where molecular structures are divided into atomic groups such as CH3 and OH, and the physical properties of an unknown structure are estimated from the contributions of the atomic groups.
*6: A method for estimating activity coefficient, solubility and other thermodynamic properties by evaluating molecular interactions in a solution from surface charge distributions obtained using quantum chemistry calculations.